Content
Help
--%>1. About ncRNAVar
ncRNAVar is a manually curated database that provides comprehensive experimentally supported associations between noncoding RNA (ncRNA) variants and human disease phenotypes. ncRNA variants have been experimentally validated as a novel class of biomarkers and potential drug targets for disease diagnosis, therapy and prognosis. The discovery of relationships between ncRNA variants and disease phenotypes has become increasingly important. ncRNAVar provides experimental validated associations between ncRNA variant-disease associations through manual curation on publications and integration of lncRNA variants from lncRNASNP and GWAS Catalog. ncRNAVar provides user-friendly web interfaces and RESTful application programming interfaces to browse, search, prioritize, analyze and access data freely.
2. The ncRNAVar web interface
The ncRNAVar database has been tested on Firefox 68.0, Google Chrome 61 and Safari.
2.1 Browse and search:
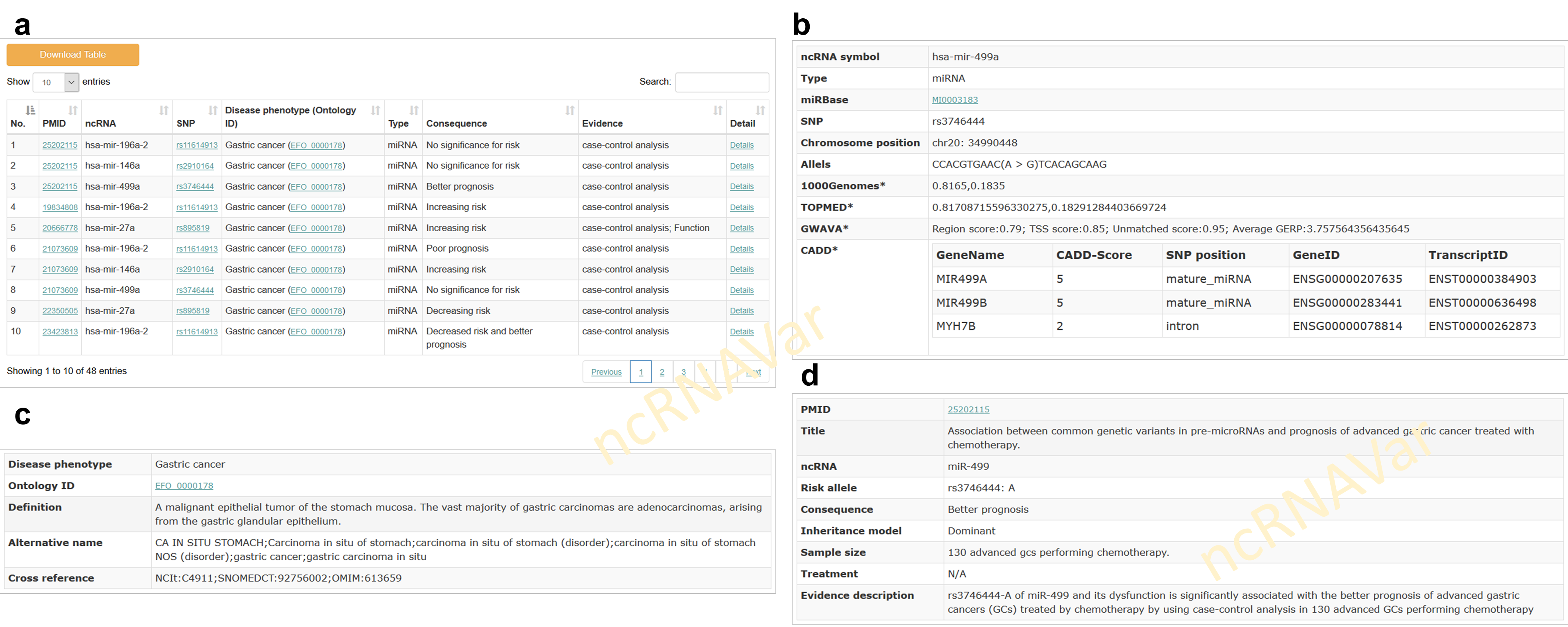
The ‘Browse’ webpage and ‘Search’ webpages in ncRNAVar allow users to quickly retrieve ncRNA variant-disease associations through searching ncRNA, variation, disease phenotype. The resulted association data is displayed in a brief table, showing key information of PubMed IDs (PMIDs), ncRNA symbols, variants, disease phenotypes with ontology identifier, the clinical consequence of the ncRNA variants, the evidence in publications (a) and the details link to further webpages for rich information of ncRNA variant (b), disease phenotype (c) and the supported evidences in publication (d). Links to other reference resources, such as PubMed, the NCBI gene, miRBase, NONCODE, piRBase, EFO, and dbSNP are also provided.
2.2 Association prioritization application:
The association prioritization application in ncRNAVar allows users to quickly retrieve a disease or ncRNA variant and promptly prioritize the ncRNA variant-disease associations. The result table allows sorting by association scores and filtering by specific disease phenotypes, ncRNAs and variants (a). The data of prioritized ncRNA variant-diseases associations can be optionally visualized in a word-cloud diagram (b and c). Larger sizes and more central locations of the ncRNA variant and disease phenotype symbols indicate higher association score between the ncRNA variants and disease phenotype.

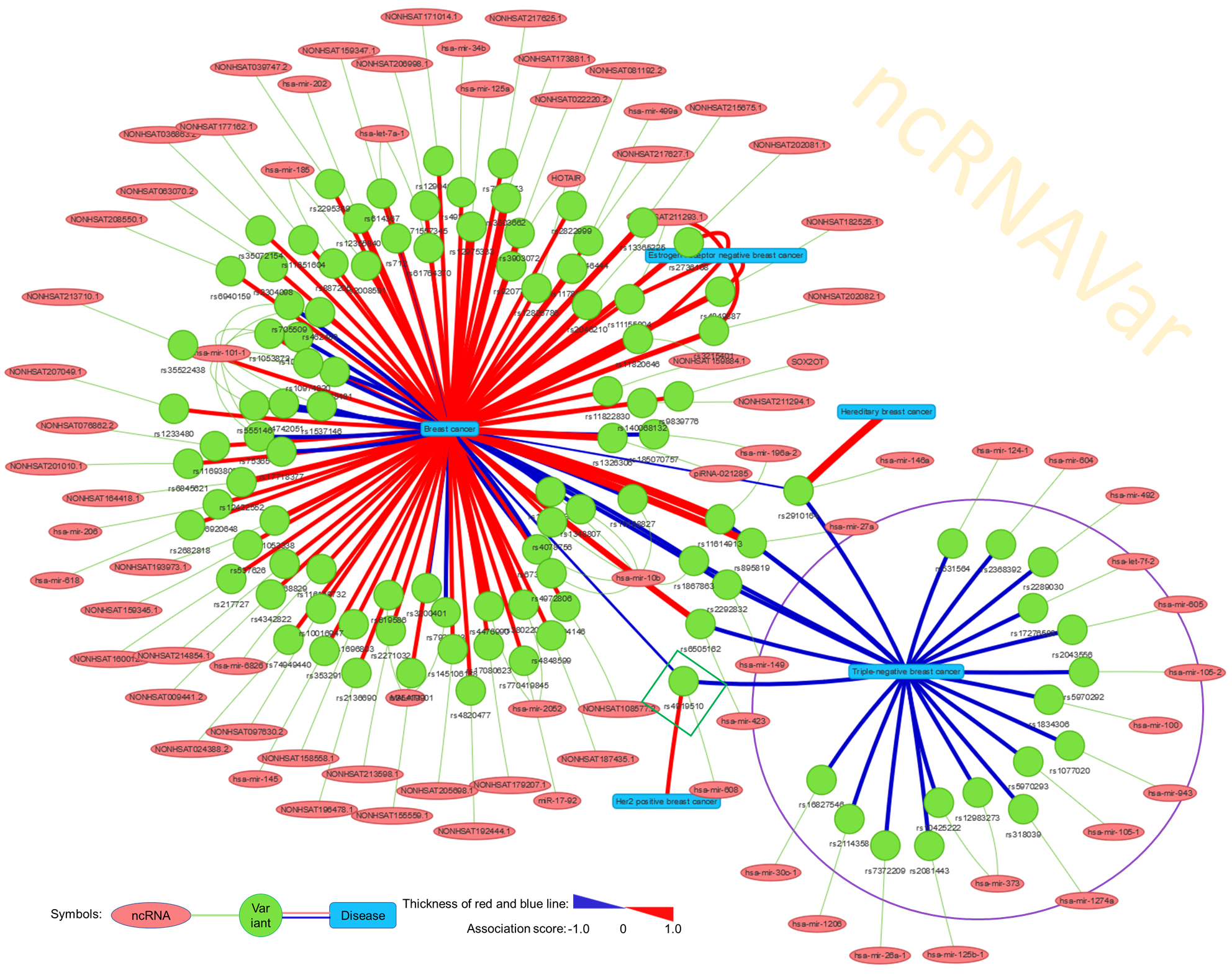
2.3 Network visualization application:
The network visualization application in ncRNAVar allows user to build and enter a set of diseases, ncRNAs or variants with a comma separation and to generate networks for overview the connections among different disease phenotypes, ncRNAs and variants. For example, we built the input of “triple-negative breast cancer,breast cancer,estrogen-receptor negative breast cancer,estrogen-receptor positive breast cancer,hereditary breast cancer,brcax breast cancer” and generated an network to overview the connections of different subtypes of breast cancer with ncRNA variants.
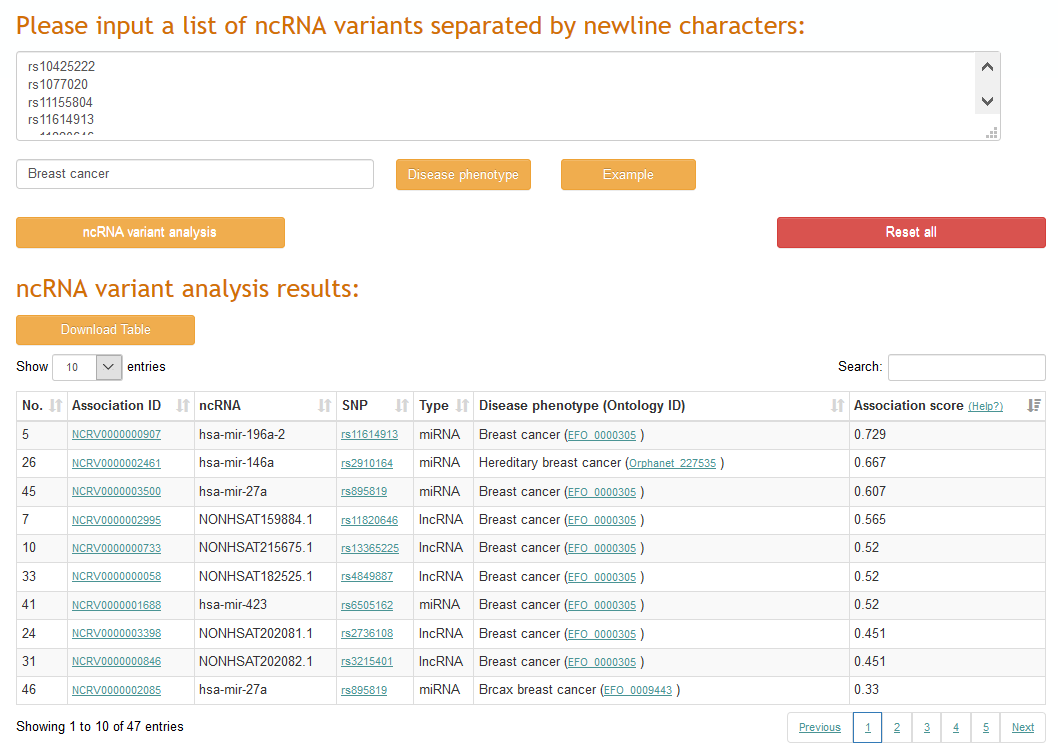
2.4 Variant mapping application:
The variant mapping application in ncRNAVar allows users to input a set of ncRNA variants with disease phenotype, and to identify the disease phenotype related ncRNA variants.
2.5 Other application interface:
ncRNAVar also offers the RESTful APIs for the programmatical access of the association data. The resulted data by the APIs are available in universal JSON and text formats. Documentation for the APIs is available on the website. All association data in ncRNAVar can be freely downloaded. In addition, ncRNAVar encourages users to submit their new association data for future data integration. Once checked by our professional curators and approved by the submission review committee, the submitted records will be included in an updated release. Furthermore, a detailed tutorial for ncRNAVar is available on the ‘Help’ webpage.
3. Computation of association score
In ncRNAVar, the diverse ncRNA variant-disease associations are manually collected and curated from different publications. Referring to the association score models in Open Targets (PMID:19107201), DisGeNET (PMID: 25877637) and MNDR (PMID: 29106639), ncRNAVar refined an association score model to prioritize the associations between ncRNA variants and disease phenotypes based on several evidential metrics. These evidential metrics include the experimental evidence types in publications (Table 1) and the number of publications.

The computation of the association score consists of three steps as below:
Step 1: In principle, validation experiments of in vivo provide more reliable evidence than in vitro such as functional and mechanism analysis. Validation experiments of functional and mechanism analysis provide more reliable evidence than genotyping analysis and prediction by using tools. Therefore, the experimentally supported evidences in publications are empirically classified into five types, and manually assigned with corresponding values, as detailed in Table 1. The evidential score of each publication (Ep) for supporting ncRNA variant-disease association is computed as below.
<%--Step 2: From a set of publications for supporting a same ncRNA variant-disease association, an overall evidential score (E) is derived using a harmonic sum function (PMID:19107201, PMID:27899665) to account for replication but also to dampen the effect of large amounts of publications by calculating:--%> Step 2: For a ncRNA variant-disease association supported or opposed by a set of publications, an overall evidential score (Es) of supporting publications and an overall evidential score (Eo) of opposed publications is derived using a harmonic sum function (PMID:19107201, PMID:27899665) to account for replication but also to dampen the effect of large amounts of supported or opposed publications by calculating respectively:
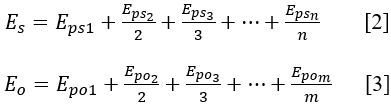
<%--As indicated in equation (2), ‘n’ represents the total number of supporting publications.--%> In equation [2], Eps1,Eps1, Eps2, Eps3, …, Epsn are the sorted evidential scores of supporting publications in descending order, and Epo1, Epo2, Epo3, …, Epom are the sorted evidential scores of opposed publications in descending order. ‘n’ and ‘m’ are the numbers of supporting publications and opposed publications, respectively. Es represents the score for significant associations and Eo represents the score for insignificant associations.
<%--Step 3: Normalizing the overall evidential score (E) to limit the range of final association scores from 0 to 1.0.--%> Step 3: Step 3: The final association scores are computed and normalized to limit the range of its from -1.0 to 1.0 as below:
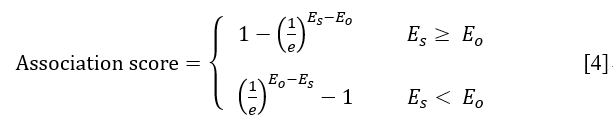
As indicated in equation (4), ‘e’ represents the natural constant e. In briefly, the association score greater than 0 indicates a significant association between the ncRNA variant and the disease phenotype, otherwise there is no significant association between the ncRNA variant and the disease phenotype.
4. Data attribute
The following information can help you to interpret ncRNA variant-disease associations.
ncRNA
To make the ncRNA symbols more consistent with other databases, ncRNAVar provides both the identifiers and links for the miRNAs in the miRBase database, the lncRNAs in the NONCODE and NCBI gene database, the piRNA in the piRBase and NCBI gene database, the snoRNAs in the HGNC and NCBI gene database, the circRNAs in the NCBI gene database.
Variant
To make the ncRNA variants more consistent with other databases, ncRNAVar provides both the identifiers and links for the ncRNA variants in the dbSNP database. ncRNAVar also provides allel frequence for the variants in 1000Genome and TOPMED. The functional impact of the variant in ncRNAVar are predicted by GWAVA and CADD tools, which are both tools for scoring the deleteriousness of single nucleotide variants as well as insertion/deletions variants in the human genome.
Disease phenotype
To make the disease phenotypes more consistent with other databases, ncRNAVar provides both the identifiers and links for the disease phenotypes in the EMBL-EBI Ontology Lookup Service resources, including Experimental Factor Ontology (EFO), Orphanet Rare Disease Ontology (ORDO), Human Phenotype Ontology (HPO), Human Disease Ontology (HDO) and NCI Thesaurus OBO Edition (NCIT) etc. The majority of diseases phenotypes in ncRNAVar are annotated by EFO, while the rest disease phenotypes, which EFO does not cover, are annotated by other ontologies.
5. Contact us
If you have any inquiries, please do not hesitate to contact us via email: liweizhong@mail.sysu.edu.cn or zhangwl25@mail2.sysu.edu.cn
